Muenkes sindroms raksturo koronāro šuvju kraniosiostoze, kas rodas FGFR3 gēna mutācijas dēļ. Slimība tiek mantota autosomāli dominējošā veidā, un to bieži pavada patoloģiskas ekstremitātes. Ārstēšana parasti atbilst ķirurģiskai procedūrai.
Kas ir Muenkes sindroms?

© flashmovie - stock.adobe.com
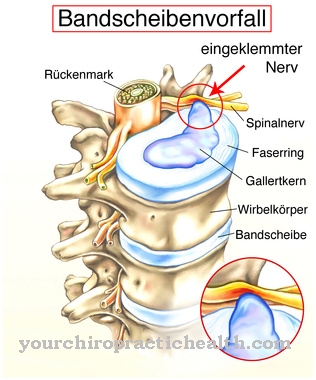
Kraniosinostozes gadījumā viena vai vairākas galvaskausa šuves embrionālās attīstības laikā priekšlaicīgi ossificējas un tādējādi novērš galvaskausa un smadzeņu fizioloģisko augšanu. Daudzas slimības no iedzimtu kroplību sindromu grupas, kurās galvenokārt tiek iesaistīta seja, satur šādas kraniosiostozes. Tāda slimība tā ir Koronāro šuvju sinostozes sindroms, ko sauc arī par Muenkes sindroms ir zināms.
Pirmoreiz slimība tika aprakstīta 1997. gadā. M. Muenke un kolēģi ir pirmie, kas to aprakstīja. Muenkes sindromu raksturo koronāro šuvju kraniosinostoze, un tas ietver arī skeleta izmaiņas tarsā un karpālos kaulos. Sindroma izplatība pašlaik nav zināma. Izpausme notiek agrīnā jaundzimušā periodā vai vēlākais mazuļu agrīnā vecumā. Lai gan slimība nav pārliecinoši izpētīta, tagad iemesls ir noskaidrots.
cēloņi
Daudzos gadījumos Muenkes sindroms varētu būt saistīts ar ģimenes uzkrāšanos. Šajos gadījumos mantojums vistuvāk atgādina autosomāli dominējošo mantojumu. Tomēr ir dokumentēti arī gadījumi, kad sindroms šķita sporādiski. Šķiet, ka iemesls ir ģenētiska mutācija, kas sporādiskos gadījumos, iespējams, atbilst jaunai mutācijai. Tiek uzskatīta arī identificēta mutācijas vieta.
Tiek apgalvots, ka slimība ir pamatota ar FGFR3 gēna mutācijām, kuras var atrast gēna lokusā 4p16.3. Arī citi sindromi ir saistīti ar FGFR3 gēnu. Viens piemērs tam ir tā dēvētais Apert sindroms. Gēns kodē DNS fibroblastu augšanas faktora receptoru 3. Maz ir zināms par FGF-3 augšanas faktora fizioloģisko iedarbību.
Saskaņā ar spekulācijām FGF-3 ir izšķirošs faktors, jo īpaši embrionālā periodā. Iespējams, ka receptoru mutācija nozīmē, ka augšanas faktors embrionālās attīstības laikā nepietiekami saistās.
Simptomi, kaites un pazīmes
Muenke sindroma pacienti cieš no dažādiem simptomiem. Sakarā ar priekšlaicīgu koronāro šuvju aizvēršanu skartajiem ir neparasta galvas forma, kas ir pamanāma arī sejas anomālijās. Papildus saīsinātajam galvaskausa priekšējam un aizmugurējam diametram acu kontaktligzdas parasti ir mazāk dziļas.
Bieži vien šie simptomi ir saistīti ar augšžokļa hipoplāziju. Ja koronārā šuve ir aizvērta vienā pusē, acu kontaktligzdas ir saplacinātas attiecīgajā pusē. Parasti sindroms neietekmē pacienta intelektu. Uz ekstremitātēm tiek atrasti roku kaulu vai tarsālo kaulu saplūšana. Nepareiza sadalīšana ir iedomājama arī uz karpālā kaula kauliem.
Kūņu epifīzes ir arī viens no iespējamiem simptomiem. Dažos gadījumos pacienta klīniskais attēls ir saistīts arī ar osteohondromiem. Fenotipiski un tādējādi simptomātiski pārklājas ar citiem sindromiem, piemēram, Pfeiffera sindromu, Džeksona-Veisa sindromu vai Sētras-Chotzen sindromu, ir klīniski domājamas izpausmes.
Diagnoze un slimības gaita
Muenkes sindroma diagnoze parasti tiek veikta jaundzimušā periodā, jo slimību var savlaicīgi atpazīt, izmantojot vizuālu diagnozi. Apmēram viens pacients no 15 000 jaundzimušajiem cieš no koronāro šuvju sinostozes. Tomēr šai parādībai nav jābūt automātiski Muenkes sindroma dēļ.
Tāpēc diagnostikai ir nepieciešams noteikt patogēno mutāciju FGFR3 gēnā. Pacienta rokas un kājas var šķist radioloģiski normālas, tāpēc diagnozes noteikšanai šajā sakarā nav pietiekami meklēt novirzes. Principā visiem bērniem ar koronāro sinostozi var veikt specifiskas P250R mutācijas skrīningu.
Šī pārbaude atbilst molekulārā ģenētiskajai analīzei. Mutācijas izslēgšana nebūt nenozīmē, ka pacients necieš no Muenkes sindroma. Dažos gadījumos mutāciju nevarēja atklāt skartajiem. Tomēr pierādījums tiek uzskatīts par drošu diagnozes noteikšanai. Sievietes prognoze nav tik labvēlīga.
Komplikācijas
Muenkes sindroma rezultātā skartie cieš no dažādām kroplībām un kroplībām, kas galvenokārt rodas uz pacienta galvas un sejas. Šīs kroplības bieži izraisa psiholoģiskas sūdzības un depresiju. Skartās personas bieži cieš no mazvērtības kompleksiem un pazeminātas pašnovērtējuma.
Simptomi bieži izraisa kauna sajūtu, un jo īpaši bērnus var ietekmēt iebiedēšana un ķircināšana Muenkes sindroma dēļ. Tomēr intelekts netiek traucēts, tāpēc pacienta garīgā attīstība norit bez komplikācijām. Pastāv arī ekstremitāšu kroplības, tāpēc ikdienas dzīvē var būt ierobežojumi dažādās aktivitātēs vai pārvietošanās ierobežojumi.
Dzīves kvalitāti ievērojami samazina Muenkes sindroms. Muenkes sindroma ārstēšanā nav citu komplikāciju. Parasti cēloņsakarības ārstēšana nav iespējama, taču, lai izvairītos no turpmākiem izrietošiem bojājumiem, jāveic dažas ķirurģiskas iejaukšanās.
Attiecīgās personas dzīves ilgums parasti nav ierobežots. Ārstēšana var notikt arī tūlīt pēc dzemdībām. Dažos gadījumos bērnu vecākus ietekmē arī Muenkes sindroma psiholoģiskās sūdzības.
Kad jāiet pie ārsta?
Raksturīgās anomālijas galvā un sejā ir skaidras Muenkes sindroma pazīmes un parasti noved pie diagnozes tūlīt pēc piedzimšanas. Vieglu simptomu gadījumā par visiem simptomiem jāinformē atbildīgais ārsts. Reizēm karpālie vai tarsālie kauli var izaugt kopā, kā rezultātā rodas slimības gaita. Cilvēkiem, kuriem jau ir šīs slimības ģimenes anamnēze, ģenētiskais tests jāveic agrīnā stadijā. Tad tūlīt pēc dzemdībām var sākt nepieciešamos ārstēšanas posmus.
Skarto bērnu vecākiem jāinformē arī ģimenes ārsts par jauniem bērna simptomiem un citām novirzēm. Turklāt vienmēr ir nepieciešama rūpīga speciālista pārbaude. Ārstēšana parasti notiek slimnīcā vai ģenētisko slimību speciālistu klīnikā. Papildus ģimenes ārstam var piezvanīt arī ortopēds vai internists. Hronisku sūdzību gadījumā terapija un fizioterapija ir arī ārstēšanas daļa.
Terapija un ārstēšana
Cēloņsakarība nav pieejama pacientiem ar Muenkes sindromu. Gēnu terapijas pieejas piedāvā cerību uz cēloņsakarības terapiju, taču tās vēl nav sasniegušas klīnisko fāzi. Ārstēšana ir tikai simptomātiska un tādējādi ir atkarīga no simptomiem katrā konkrētajā gadījumā. Lai labotu galvaskausa anomālijas, ir iespējamas tikai ķirurģiskas ārstēšanas iespējas.
Operācija ir paredzēta, lai palīdzētu mazināt spiedienu uz smadzenēm, ko izraisa koronāro šuvju agrīna aizvēršana. Galvas nervi šādā veidā tiek atbrīvoti, un pēc procedūras tie ir ideāli saspiesti vai nemaz nav saspiesti. Ir pieejama konservatīva ārstēšanas iespēja mazāk smagām kraniosinostozēm. Piemēram, bērniem, kurus vieglāk ietekmē, var piešķirt galvaskausa formas, kuras viņiem ilgi jāvalkā.
Šīs galvaskausa formas mēģina adekvāti modelēt galvaskausu. Tā kā Muenke sindroms parasti tiek diagnosticēts jau jaundzimušajam, šāda konservatīva rekonstruēšana ir īpaši noderīga: zīdaiņu galvas forma joprojām ir pielāgojama. Konservatīvajai modelēšanai galu galā ir tāds pats mērķis kā operatīvajai modelēšanai.
Ārstēšana nodrošina smadzeņu fizioloģisko augšanu. Turklāt galvas izskats tiek pielāgots vidējam. Vienlaicīgus simptomus, piemēram, ekstremitāšu kroplības, var ārstēt ķirurģiski. Ja tie neierobežo un netraucē skarto personu, šāda ārstēšana nav absolūti nepieciešama.
Perspektīva un prognoze
Muenkes sindroms ir reta slimība, kuru tagad var ārstēt ķirurģiski. Ja savlaicīgi tiek atklāta koronāro šuvju priekšlaicīga aizvēršanās, prognoze ir laba. Jebkuras kroplības var ārstēt ķirurģiski. Piemērotus medikamentus var izrakstīt tādiem simptomiem kā sāpes vai jutīguma traucējumi. Koronāro šuvju sinostozes sindroms nesamazina paredzamo dzīves ilgumu.
Dzīves kvalitāti var nedaudz ierobežot ar minētajām sūdzībām, kā arī iespējamām rētām uz sejas un galvas. Tomēr kopumā prognoze ir pozitīva. Bērni ar Muenkes sindromu ir īpaši pakļauti iekšējās auss dzirdes zudumam. Slikta dzirde vēlāk var radīt problēmas, piemēram, kad pacients vairs nespēj saprast instrukcijas vai atrast ceļu ikdienas dzīvē.
Izslēgšanas un samazinātas pašnovērtējuma dēļ dažiem slimniekiem rodas psiholoģiskas sūdzības. Labklājību var samazināt, ja slimniekiem nav laba atbalsta tīkla vecāku, radinieku, draugu un terapeitu veidā. Bērniem, kuri cieš no Muenkes sindroma, ikdienas dzīvē noteikti ir nepieciešams atbalsts, lai, neskatoties uz slimību, viņi būtu garīgi un fiziski veseli.
novēršana
Muenkes sindromu nevar novērst, jo to ietekmē nevis ģenētiski, bet gan ģenētiski faktori. Ģenētiskās konsultācijas grūtniecības laikā ir vienīgais profilakses līdzeklis.
Pēcaprūpe
Vairumā gadījumu tiem, kurus skārusi Muenkes sindroms, tiešajai sekojošajai aprūpei ir pieejami ļoti maz un tikai ļoti ierobežoti pasākumi. Pirmkārt un galvenokārt, ir ātri un, galvenais, agri diagnosticējot šo slimību, lai turpmāk nerastos komplikācijas. Tā kā Muenke sindroms ir ģenētiska slimība, to nevar pilnībā izārstēt.
Tomēr, ja vēlaties bērnus, ģenētiskā pārbaude un konsultācijas var būt noderīgas, lai novērstu sindroma atkārtošanos. Lielākā daļa no skartajiem ir atkarīgi no operācijas. Pacientam noteikti vajadzētu atpūsties un pēc šādas operācijas to ņemt viegli, ideālā gadījumā būtu jāsaglabā gultas režīms. Jāizvairās arī no nogurdinošām vai fiziskām aktivitātēm, lai nevajadzīgi neradītu ķermenim stresu.
Daudzos gadījumos sindroma skartie ir atkarīgi arī no savas ģimenes palīdzības un atbalsta ikdienas dzīvē. Mīlošas un intensīvas sarunas ar ģimeni un draugiem arī ļoti pozitīvi ietekmē turpmāko slimības gaitu. Tas nav nekas neparasts, lai novērstu depresijas vai citu psiholoģisku traucējumu attīstību.
To var izdarīt pats
Muenkes sindromu parasti ārstē ķirurģiski un ar medikamentiem. Vissvarīgākais pašpalīdzības pasākums ir rūpēties par ķermeni pēc procedūras un cieši konsultēties ar atbildīgo ārstu. Skarto bērnu vecākiem uzmanīgi jānovēro bērns un, ja rodas novirzes, jāinformē atbildīgais ārsts.
Kopumā Muenkes sindromu var ārstēt salīdzinoši labi, un tas nerada pastāvīgus dzīves kvalitātes traucējumus. Tomēr kroplības un kroplības gandrīz vienmēr paliek, kas dažreiz var būt būtisks emocionālais slogs skartajiem. Šī iemesla dēļ medicīniskā ārstēšana ir jāatbalsta ar terapeitiskiem pasākumiem.
Smagi cietušajiem bērniem ar attīstības atšķirībām agrāk tika ieteikts apmeklēt speciālo bērnudārzu un vēlāk speciālo skolu. Tā vietā šodien tiek atsaukta uz iekļaušanu vispārizglītojošajās skolās. Bērni bez jebkādām citām novirzēm jebkurā gadījumā var apmeklēt parasto un augstāko skolu.
Ir nepieciešama visaptveroša terapija, īpaši smagu mobilitātes ierobežojumu gadījumā. Vecāki var atbalstīt šos pasākumus, atbalstot bērnu ikdienas dzīvē. Parasti ir norādīti turpmāki fizioterapeitiskie pasākumi.
Tā kā slimība var radīt ievērojamu slogu arī radiniekiem, vecākiem un draugiem jāmeklē arī terapeitiskā palīdzība. Terapeits var arī nodibināt kontaktu ar citām skartajām personām un vajadzības gadījumā vecākus novirzīt pašpalīdzības grupā.


.jpg)











.jpg)