Ušera sindroms ir dažādu hromosomu gēnu mutācija, kas izraisa dažāda smaguma pakāpes dzirdes un redzes traucējumus. Vissmagākā forma ar iedzimtu kurlumu un redzes lauka zaudēšanu ir saistīta ar desmit gadu vecumu. Kaut arī daļēji progresējošos dzirdes traucējumus ārstē ar dzirdes aparātu un vēlāk ar cholelear implantu, redzes lauka zuduma ārstēšanai nav pieejamas terapijas metodes. Tomēr zinātnieki šobrīd pēta cilmes šūnu terapijas un tīklenes implantu iespēju.
Kas ir Ušera sindroms?
Ušera sindroms ir iedzimta redzes pasliktināšanās, ko izraisa dažādas hromosomu mutācijas. Pusi no visiem 40 sindromiem dzirdes un redzes akluma jomā var izsekot Ušera sindromam. Šīs parādības sastopamība ir no trim līdz sešiem no 100 000 gadījumiem, vairums no tiem notiek sestajā dzīves desmitgadē. Neziņoto slimības gadījumu skaits, iespējams, ir krietni lielāks par pieminēto.
Tā kā Ušera sindroms bieži tiek sajaukts ar parasto dzirdes zudumu, savukārt tā saistība ar pavadošo aklumu, ko izraisa izplatītā tīklenes slimība, pigmentosa retinīts, bieži netiek atzīta pietiekami agri. Pirmais sindroma apraksts ir meklējams Albrehta fon Grēfera 19. gadsimtā, taču tajā laikā tas nebija pietiekami diferencēts un dokumentēts. Britu oftalmologs Šarls Hovards Ušers vēlāk tika nosaukts par šo fenomenu, jo viņš pirmo reizi paziņoja par sindromu un šajā jomā veica novatorisku darbu.
cēloņi
Ušera sindromu izraisa dažādas gēnu mutācijas, kuras visas ir iedzimtas. 11. hromosomu, 17. hromosomu un 3. hromosomu mutācija īpaši ietekmē. Lai arī klīniski nav nozīmes tam, kura no hromosomām mutē, ārsts diferencē Usher sindromu dažādos apakštipos starp 1B un 3B atkarībā no ietekmētajām hromosomām.
Atkarībā no apakštipa mutācijas forma un līdz ar to arī faktiskais izskata cēlonis ir atšķirīgi. Pa to laiku zinātne balstās uz tā saucamajiem Usher olbaltumvielu kompleksiem, kas katrs ietekmē dažādu olbaltumvielu izvietojumu membrānā. Domājams, ka šie proteīni ir iesaistīti redzes un dzirdes maņu šūnu signālu pārraidē. Tātad, ja trūkst šo kompleksu viena komponenta, viss olbaltumvielu komplekss šūnā nevar veikt savus faktiskos uzdevumus, un tā laikā attiecīgās šūnas deģenerējas. Pirmām kārtām svarīgu lomu šajā sakarā vajadzētu būt Usher 1C apakštipa sastatņu olbaltumvielu harmonikai.
Simptomi, kaites un pazīmes
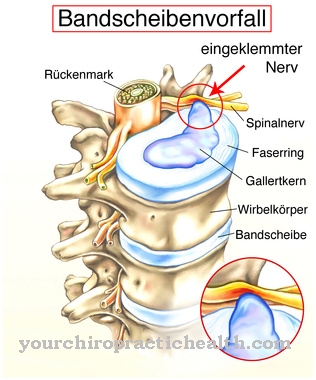
Kā parasti, Ušera sindroms pirmajos gados izpaužas kā iekšējās auss dzirdes zudums, kura pamatā ir bojājumi matu šūnās cochlea. Retāk nekā tikai dzirdes zudums ir pilnīgs kurlums no dzimšanas brīža.Papildus šiem agrīnajiem simptomiem, tīklenes slimība retinopathia pigmentosa ir viens no Ushera sindroma simptomiem, kas rodas vēlāk.
Šīs slimības gadījumā fotoreceptori pamazām mirst no perifērijas līdz makulai. Nakts sākumā tiek iestatīts aklums. Vēlāk redzes lauks ir ierobežots un tā sauktā tuneļa redze. Atkarībā no apakštipa šī parādība var izraisīt pilnīgu aklumu. Dažos gadījumos šos simptomus papildina arī epilepsijas lēkmes.
Diagnoze un slimības gaita
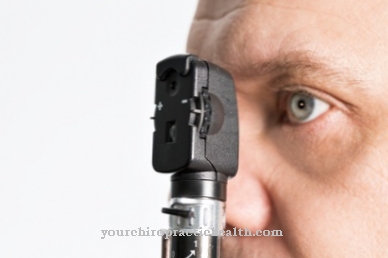
Usher sindroma diagnozi ārsts veic, izmantojot dažādas diagnostikas metodes. Diagnoze pēc iespējas agrāk ir ļoti svarīga, jo pretējā gadījumā pacientam nebūs laika garīgi un fiziski pielāgoties vēlākam maņu uztveres zaudējumam un, iespējams, sagatavoties, apgūstot jaunas komunikācijas metodes.
Papildus elektroretinogrammai var notikt DNS mikroshēma, olbaltumvielu mikroshēma vai ģenētiskā analīze. Slimības gaita ir ļoti atkarīga no veida. Atkarībā no slimības smaguma var izdalīt trīs veidus. 1. tipa uztvērējs (USH1) atbilst vissmagākajai slimības gaitai, kurā ir kurlums no dzimšanas brīža un dažreiz traucēta līdzsvara izjūta.
Retinopathia pigmentosa sākas šajā apakštipā no desmit gadu vecuma. 2. tipu Usher (USH2) nosaka ar pastāvīgu, dziļu dzirdes zudumu, pubertātes laikā sākot ar pigmentosa retinopathia. Ar Usher tipa 3 (USH3) progresējošs dzirdes zudums un redzes lauka zudums sākas tikai dzīves otrajā pusē.
Komplikācijas
Ušera sindroma dēļ skartās personas cieš no ļoti nopietnām kaites un simptomiem. Parasti dzirdes zudums rodas ļoti jaunā vecumā. Īpaši jaunieši cieš arī no psiholoģiskām sūdzībām vai pat no depresijas.
Turpmākajā kursā beidzot ir pilnīgs dzirdes zudums un tādējādi ievērojami ierobežojumi ikdienas dzīvē. Bērniem Ušera sindroms arī noved pie aizkavētas un ļoti ierobežotas attīstības. Slimība var izraisīt arī nakts aklumu. Retos gadījumos skartie cieš no pilnīgas akluma.
Sindroms izraisa arī epilepsijas lēkmes, kas ir saistītas ar ļoti stiprām sāpēm. Sliktākajā gadījumā tas var izraisīt arī attiecīgās personas nāvi. Daudzi pacienti ir atkarīgi no citu cilvēku palīdzības, ņemot vērā sūdzības ikdienas dzīvē, un paši nevar tikt galā ar ikdienas dzīvi.
Ar dažādu terapiju palīdzību dažus simptomus var mazināt. Tomēr slimības progresēšanu nevar ierobežot. Ikdienas pacienti ir atkarīgi no dzirdes un redzes palīglīdzekļiem. Ušera sindroms nemaina dzīves ilgumu.
Kad jāiet pie ārsta?
Tā kā Ušera sindroms pats nevar dziedēt, skartā persona ir atkarīga no ārsta apmeklējuma. Tas ir vienīgais veids, kā novērst turpmākas komplikācijas vai sūdzības, kas apgrūtina skartās personas dzīvi. Tā kā tā ir ģenētiska slimība, to nevar pilnībā izārstēt. Tomēr, ja vēlaties bērnus, var veikt ģenētiskās konsultācijas, lai Ušera sindroms pēcnācējos neatkārtotos. Ja cietušais bērns ir pilnīgi kurls, ar šo slimību jākonsultējas ar ārstu. Tas ir bijis kopš dzimšanas.
Turklāt acīs ir arī diskomforts, kas arī pasliktinās ar vecumu. Pacienti cieš arī no nakts akluma, kas var norādīt arī uz sindromu. Dažos gadījumos Ušera sindroms izpaužas ar epilepsijas lēkmēm. Šāda uzbrukuma gadījumā nekavējoties jāizsauc neatliekamās medicīniskās palīdzības ārsts vai tieši jāapmeklē slimnīca. Pašu sindromu var noteikt ģimenes ārsts vai pediatrs. Turpmāka ārstēšana ir atkarīga no simptomu nopietnības, un to veic speciālists. Parasti šis sindroms nesamazina pacienta dzīves ilgumu.
Ārstēšana un terapija
Pašlaik Ushera sindromā nav retinopathia pigmentosa ārstēšanas. Pašlaik tiek pētītas gēnu terapijas pieejas, un tām drīz vajadzētu ļaut aizvietot bojātos gēnus tīklenē. Turklāt pašlaik tiek pētītas tādas terapijas iespējas kā cilmes šūnu terapija un tīklenes implanti.
Ushera sindroma dzirdes traucējumus ārstē ar dzirdes aparātu, un vēlākos posmos var būt nepieciešams košlera implants. Parasti pacienti ar Ušera sindromu agrīni apgūst zīmju valodu. Ja rodas vai pastāv nopietni redzes lauka traucējumi, lai saglabātu komunikācijas prasmes, var iemācīties taktilo zīmju valodu. Jūs ņemat otra cilvēka rokas sevī un jūtat attiecīgos žestus.
novēršana
Tā kā Ušera sindroms ir iedzimta slimība, kuras pamatā ir gēna mutācija, šo parādību nevar novērst. Tā kā šai slimībai ir svarīga agrīna diagnostika, regulāra paša dzirdes un redzes lauka īpašību kontrole ir vismaz viens pasākums, kas, izmantojot agrīnu diagnozi, var palielināt cilvēka spēju pielāgoties mainītajiem apstākļiem.
Pēcaprūpe
Ušera sindroms ir dažādu hromosomu gēnu mutācija, kas parasti ir saistīta ar dzirdes zudumu un smagiem redzes traucējumiem. Cietējiem ir jāvalkā dzirdes aparāts; kohleārs implants var palīdzēt ar ļoti smagu dzirdes zudumu. Slimam cilvēkam, kurš kopš dzimšanas ir cietis no Ušera sindroma, ikdienas dzīvi parasti ir vieglāk pārvarēt. Pacienti, kuri dzīves laikā kļūst nedzirdīgi un, iespējams, ir akli, bieži ir daudz problemātiskāki slimības apkarošanā.
Jūsu ikdienas dzīve pēkšņi ir ļoti ierobežota un stresa stāvoklī. Tāpēc ļoti svarīgs ir paziņu, draugu, speciālistu un atbilstošu terapeitu atbalsts. Izmantojot optimālu ārstēšanu, skartajai personai bieži ir iespējams dzīvot normālu dzīvi. Palīdzība ikdienas dzīvē, protams, joprojām ir nepieciešama: Personas dzīvoklim jābūt pieejamam ar invaliditāti. Ja pacientam ir darbs, tam jāatbilst viņa fiziskajām iespējām, un darba vieta jāpielāgo viņa invaliditātei.
Jānodrošina kontakts ar citiem cilvēkiem, lai skartie cilvēki pilnībā neatkāpjas. Saziņa var notikt, izmantojot lūpu lasīšanu vai zīmju valodas. Ja slimības gaitā palielinās aklums, saziņai var izmantot arī tā saukto taktilo zīmju valodu, atbilstošās pazīmes tiek nodotas ķermeņa kontakta ceļā. Ushera sindroma asociācija piedāvā kompetentu kontaktpunktu skartajiem.
To var izdarīt pats
Ušera sindroms prasa ārstēšanu. Cietušajiem ir jāvalkā dzirdes aparāts vai, ja viņiem ir smagi dzirdes zudumi, implants, lai uzlabotu dzirdi.
Cilvēkiem, kuri no dzimšanas cieš no Ušera sindroma, parasti ir vieglāk. Pacientiem, kuri dzīves laikā kļūst tikai nedzirdīgi un beidzot ir akli, pēkšņi ierobežojumi rada lielu slogu ikdienas dzīvē, tāpēc ir svarīgi saņemt labu draugu, paziņu, speciālistu un terapeitu atbalstu. Tad pacientam var būt iespējama optimāla ārstēšana, caur kuru var dzīvot salīdzinoši normālu dzīvi.
Tomēr cilvēki, kuri ir nedzirdīgi un neredzīgi, ir atkarīgi no atbalsta ikdienas dzīvē. Dzīvoklim jābūt aprīkotam ar invaliditāti, un darbam jāatbilst paša fiziskajām spējām. Ietekmētajiem jau agri jāapgūst lūpu lasīšana un citas metodes, lai varētu labāk komunicēt ar citiem cilvēkiem ikdienas dzīvē. Palielinoties aklumam, saziņai var izmantot arī tā saucamo taktilo zīmju valodu. Iespējamās valodas ir, piemēram, Tadoma vai Gestuno, pēdējās koncentrējas uz zīmju sajūtu, nonākot ķermeņa kontaktā. Ušera sindroma asociācija V. piešķir skartajiem papildu kontaktpunktus.

.jpg)

.jpg)










.jpg)