Hipoksantīns ir purīna novirze un nonāk saistītā formā kā nukleobāze un brīvā formā, piem. B. urīnā. Tas atrodams arī dziedzeros un kaulu smadzenēs. Kā adenīna dezaminācijas produkts, hipoksantīns tiek oksidēts līdz urīnskābei un ksantīnam. Retāk tas veido nukleīnskābju pamata struktūru.
Kas ir hipoksantīna guanīna fosforibosiltiltransferāze?
Tetrameriskais enzīms veidojas no hipoksantīna un guanīna Hipoksantīna guanīna fosforibosiltransferāze.
Tetramers ir makromolekulas, kas sastāv no četriem līdzīgiem celtniecības blokiem, precīzāk no monomēriem. Enzīms ir viens no vissvarīgākajiem eikariotu purīna metabolismā, tas ir jutīgs pret gēnu izmaiņām un var izraisīt novirzes cilvēkiem caur gēnu mutācijām, kas tiek izteiktas noteiktās vielmaiņas slimībās. Tādi ir z. B. Leša-Nīhana un Kellija-Seegmillera sindromi.
Funkcija, efekts un uzdevumi
Ferments hipoksantīna-guanīna-fosforibosiltransferāze palielina purīna metabolismu un tā enerģētisko efektivitāti.
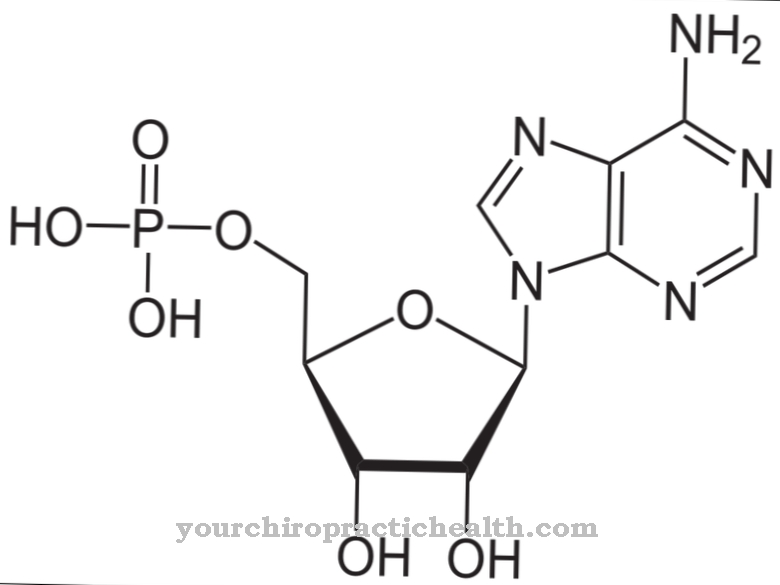
Tās pamatā ir purīna bāzes, kas ir nukleīnskābes, kuras strukturāli iegūtas no purīna. Tie ir ksantīns, hipoksantīns, adenīns un guanīns, kas ar ūdeņraža saitēm saistās ar citām bāzēm. Šādām saitēm ir liela ietekme uz DNS dubulto spirāli un replikāciju, un tām ir loma olbaltumvielu biosintēzē.
Purīna bāzes var pārstrādāt divi fermenti. Papildus hipoksantīna guanīna fosforibozil-transferāzei šī ir arī adenīna fosforibosil-transferāze. Abi veido nukleotīdu caur fosforibosilgrupas atlikumu, kas savukārt ir nukleīnskābju pamata celtniecības bloks gan DNS, gan RNS. Molekulā ir cukurs, bāze un fosfāts, un tā kontrolē dzīvībai svarīgās regulatīvās funkcijas šūnās. Veidošanās laikā tiek saglabāts ATP un samazināta urīnskābes veidošanās.
Ja purīna bāzes tiek pārstrādātas, to sauc par glābšanas ceļu. Šis ir vispārīgs termins vielmaiņas ceļiem, kuros biomolekulu sintēze rodas noārdīšanās produktiem. Organisms pats veic pārstrādes procesu, kurā aptuveni deviņdesmit procenti purīnu bāzu tiek atkārtoti izmantoti un desmit procenti faktiski tiek izvadīti. Tas parāda purīna bāzes pārstrādes efektivitāti un hipoksantīna-guanīna fosforibosiltransferāzes nozīmi.
Izglītība, sastopamība, īpašības un optimālās vērtības
Ja mutācijā notiek HPRT gēns, lielums un aminoskābes var mainīties. Tā var būt papildu DNS sekvenču vai nukleotīdu iekļaušana, kas savukārt noved pie nepareiza attiecīgā gēna kodētā gēna produkta ražošanas vai pat visas sekvences izdzēšanas. Vai z. B. maina aminoskābju secību, rodas tādas slimības kā podagra.
Īpaši nopietnas ir metabolisma slimības, piemēram, Leša-Nyhana sindroms ģenētiska defekta rezultātā. Tas tiek mantots ar x-saistīta recesīvā veidā, kas nozīmē, ka tas galvenokārt ietekmē vīriešus, kuriem ir tikai viena X hromosoma. Ģenētiskais defekts var būt sievietēm, bet tas izdalās tikai kā slimība, kad tiek skartas abas X hromosomas, kas ir samērā reti. Visbiežāk otrā X hromosoma kompensē pirmās defektu.
Jūs varat atrast savus medikamentus šeit
➔ Zāles urīnpūšļa un urīnceļu veselībaiSlimības un traucējumi
Sindroms izpaužas kā hipoksantīna guanīna fosforibosiltransferāzes deficīts. Fermentu neražo ģenētiskā defekta dēļ. Guanīna un hipoksantīna bāzu mutāciju un atkārtotas pārstrādes un pārveidošanas trūkuma dēļ uzkrājas purīna bāzes, kuras ķermenim ir jāveido un jāizvada.
Sadalīšana notiek caur starpproduktu ksantīnu, kas tiek pārveidots par urīnskābi un izdalās caur nierēm. Ja šis process tiek ierobežots, locītavu zonā veidojas urīnskābes kristāli, kas pēc tam izraisa vēl vairāk podagras lēkmes. Fermentu vairs neražo, paaugstinās urīnskābes līmenis audos un asinīs, tiek traucēta centrālā nervu sistēma.
Lesch-Nyhan sindroms nav tieši redzams dzimšanas brīdī. Kāju pamanāmo stāvokli un bērna tieksmi nedaudz kustēties un lēnāk attīstīties var redzēt tikai pēc apmēram desmit mēnešiem. Sindroms ir vājš un smags. Palielināta urīnskābes sekrēcija un vieglāki podagras lēkmes ir maigāka forma, ar smagiem simptomiem ir paškaitējums, smagi garīgi traucējumi un agresija. Paškaitējums notiek ar pirkstu vai lūpu kodumiem. Iekojot ekstremitātēs, bieži var novērot, ka skartie cilvēki ierobežo savu autoagresiju tikai ar vienu roku. Savukārt agresija bieži tiek vērsta pret jums tuviem cilvēkiem, piemēram, brāļiem un māsām vai vecākiem.
Visnopietnākajai slimības formai raksturīgas vairākas neiroloģiskas disfunkcijas un ļoti izteikta tieksme uz pašsakropļošanos. Sindroms izpaužas kā spastiskums, distonija, hipotonija, horeoatetoze un pastiprināta vēlme reaģēt uz refleksiem. Psihiskās īpašības un attīstība ir stipri ierobežota. Šajā stāvoklī sindroms var izraisīt nāvi arī īpaši drastiskā mērā.
Slimība tiek diagnosticēta, izmantojot medicīnisko attēlu. Izmēra urīnskābes līmeni urīnā un asinīs, kā arī hipoksantīna-guanīna fosforibosiltiltransferāzes aktivitāti audos un asinīs. Pēdējais ir ievērojami samazināts, un tas var būt arī pirmsdzemdību periodā.
Slimības ārstēšana ir sarežģīta. Izārstēt nav iespējams, un bez ārstēšanas bērns mirst pirmajos dzīves gados. Dažos gadījumos mazuļa zobi ir jāizņem kā profilakses līdzeklis. Citas terapeitiskās pieejas ietver urīnskābes līmeņa pazemināšanu, izmantojot tādas zāles kā allopurinols, kas podagras gadījumā darbojas kā inhibitors. Purīna bāzes netiek pārstrādātas, bet urīnskābe tiek labāk sadalīta. Ārstē arī attiecīgos traucējumus, infekcijas un nervu bojājumus, un ieteicama īpaša diēta, kas parasti nesatur gaļu un kurā ir maz purīna.
Tiek veikti arī pētījumi par smadzeņu dziļas stimulācijas psihosomatiskajām blakusparādībām. Medicīna cer, ka tas novērsīs agresiju un sevis sakropļošanu. Savukārt Kellija-Seegmillera sindroms ir vieglākais hipoksantīna-guanīna fosforibosiltiltransferāzes deficīta veids. Arī šeit tiek ražots pārāk daudz urīnskābes un rodas agrīnas podagras slimības. Pirmās sindroma pazīmes ir oranžie kristāli bērna autiņā, urīnceļu infekcijas un urolitiāze. Podagra vai akūts artrīts attīstās pubertātes laikā.
Psihiska nepietiekama attīstība un pašizbrukumi, kā tie notiek ar Leša-Nīhana sindromu, neattiecas uz lietu, lielākoties tas var izraisīt uzmanības traucējumus. Ārstēšana agrīnā stadijā parasti ietekmē skartajiem cilvēkiem paredzamo dzīves ilgumu.



.jpg)


.jpg)



.jpg)

.jpg)
.jpg)