Jouberta sindroms raksturo smadzeņu stumbra iedzimta kroplība, kā arī agenesis (kavējoša kroplība, piesaistes trūkums, piemēram, smadzeņu stieņi, pielikums). Var pastāvēt arī smadzeņu tārpa hipoplāzija (nepietiekama attīstība). Pacientiem, kuri cieš no šī autosomāli recesīvā ģenētiskā defekta, cita starpā ir vērojama patoloģiska elpošanas izturēšanās un ataksija.
Kas ir Jouberta sindroms?

© Saškina - stock.adobe.com
Cilvēki ar Jouberta sindroms cieš no centrālās nervu sistēmas attīstības traucējumiem un no tiem izrietošajiem funkcionālajiem traucējumiem. Medicīniskie pētījumi ir diskutabli par to, vai šo ģenētisko traucējumu pats par sevi vajadzētu klasificēt kā slimību.
Skartiem pacientiem ir daudz dažādu simptomu. Tādēļ ir grūti noteikt galīgo diagnozi. JB raksturo plaša gēnu lokusa neviendabība. Līdz šim ir identificētas vairākas gēnu mutācijas. Mutāciju analīze ir ļoti plaša.
cēloņi
Jouberta sindroms pieder primāro ciliofātiju grupai. Ar šo ģenētisko primārā cilia vai bazālā ķermeņa traucējumu var rasties dažāda veida attīstības traucējumi. Kā īpašie šūnu procesi cilijas veic dažādus uzdevumus. Tie darbojas kā ķīmijas, mehāniskās un osmozes sensori un ir iesaistīti daudzos signalizācijas ceļos. Turklāt tie nodrošina normālu orgānu attīstību.
Viņi uztur pamata attīstības procesu audu homeostāzi. Liels skaits iesaistīto olbaltumvielu mijiedarbības rezultātā veido sarežģītu tīklu. Ja papildus galvenajiem simptomiem tiek ietekmēti arī citi orgāni, tad ir JSRD (ar Jouberta sindromu saistīts traucējums). Šo sekundāro slimību raksturo turpmākās orgānu izpausmes, kurās iesaistītas nieres, aknas un acis.
Tas ir ģenētiski neviendabīgs sindroms. Ārsti ir atraduši anomālijas NPHP6 / CEP290 gēnā (kodē nefrocistīnu-6) vai NPHP8 / RPGRIP1L gēnā (kodē nefrocistīnu-8). Citas gēnu mutācijas ir MKS3, ARL13B, AHI1, CC2DA2, TMEM216 un INPP5E. Tikai dažiem pacientiem ir mutācijas NPHP4 un NPHP1.
Simptomi, kaites un pazīmes
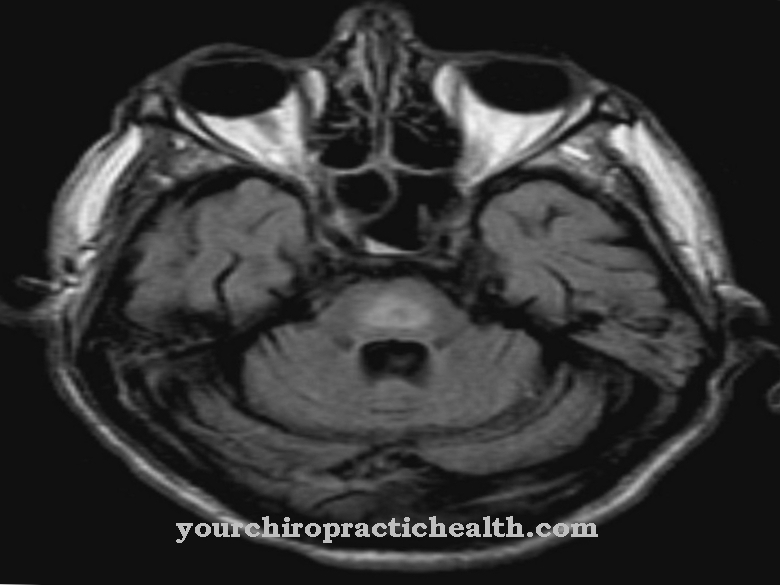
Patognomoniskā pazīme ir "molārā zoba zīme" (MTS), ko var noteikt, izmantojot "aksiālo T1 svērto smadzeņu magnētiskās rezonanses attēlveidošanu". Šo pazīmi raksturo smadzeņu vai smadzenīšu tārpu agenesis vai hipoplāzija. Turklāt aizmugurējā starpnozaru fossa (bedre starp smadzeņu kājām) ir stingri ievilkta, un smadzenīšu kātiņiem ir izteikta labāka forma vidējās smadzeņu kroplības dēļ.
Papildus MTS pacienti bieži cieš no elpošanas traucējumiem, ataksijas, muskuļu hipotensijas un psihomotoriskas atpalicības. 8 līdz 19 procentiem no skartajiem parādās postaksiāls poliaktilijs (vairāki pirksti) un sešiem procentiem pakauša (meningo) encefalocēle, kurā smadzeņu aizmugure ir izliekta.
Pirmoreiz šī kroplība tika reģistrēta 1969. gadā. Izplatība ir aptuveni 1: 100 000, attiecība parāda, cik reti slimība parādās. Kopš pirmās medicīniskās apskates ir dokumentēti tikai simts gadījumi. Tā kā šis ģenētiskais defekts rodas dažādās formās un variantos, ārsti pieņem vairākas izmaiņas ģenētikā.
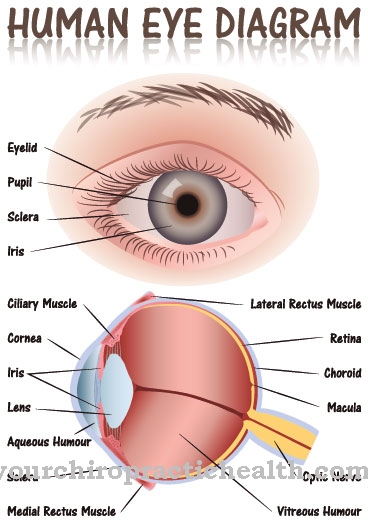
Precīza anomālija vēl nav pārliecinoši pārbaudīta. Tomēr X hromosomas mutācija tiek uzskatīta par noteiktu. Šie traucējumi tiek nodoti, pamatojoties uz autosomāli recesīvu mantojumu. Tiek iesaistīti trūkstošie vermis cerebelli (smadzenītes, smadzeņu tārps), tīklenes bojājumi un manāms varavīksnenes.
Bieži sastopami simptomi un sūdzības jaundzimušā periodā ir nistagms un neregulāra elpošanas forma kā epizodiska tahikona un apnoja. Maziem bērniem var attīstīties hipotonija. Palielinoties vecumam, attīstās nelīdzsvarotība un nevienmērīga gaita (ataksija). Šie galvenie simptomi ir zināmi arī kā motora atskaites punkti.
Pacientiem ir atšķirīgs kognitīvo spēju līmenis, un viņi var būt nopietni traucēti, taču viņi var parādīt arī normālu intelekta līmeni. Ir iespējama arī okulo-motora apraksija (kustību traucējumi).
Šim ģenētiskajam defektam raksturīgas kraniofaciālas anomālijas, piemēram, liela galva, noapaļotas un augstas uzacis, ievērojama (izvirzīta) piere, deformēta mute, ritmiski kustīga un izvirzīta mēle un dziļi novietotas ausis. Reizēm raksturīgi simptomi ir nefrofīts, tīklenes distrofija un poliaktilija.
Diagnoze un slimības gaita
Diagnoze tiek veikta, pamatojoties uz iepriekšminētajiem raksturīgajiem ataksijas, hipotensijas, okulomotorās apraksijas, atklātas vermis cerebelli pagrieziena punktiem pēc 18. grūtniecības nedēļas un attīstības kavēšanos. Turklāt MRT tiek veikts raksturīgs neiroradioloģisks atradums - MTS (molārā zoba zīme).
Šī pazīme, kas pazīstama kā molārā zīme, ir radusies kroplības krustveida un vidējā smadzenēs, kā arī mazā smadzeņu tārpa hipoplāzijas dēļ. Diferenciāldiagnozes tiek veiktas, pamatojoties uz slimībām, kas ir cieši saistītas ar JS, piemēram, JSRD (ar Jouberta sindromu saistīti traucējumi), Dandija-Walkera kroplībām (kroplīgs smadzeņu tārps bez MTS), okulomotorās apraksijas 1. un 2. tipam, ponto-smadzeņu hipoplāzijai un atrofijai, 3-c. Sindroms, orofacio-digital sindromi II un III, kā arī Mekela-Grūbera sindroms.
I stadijā ietilpst gēnu JBTS5 (53 kodējošie eksoni), JBTS3 (26 kodējošie eksoni), JBTS6 (28 kodēšanas eksoni) un JBTS9 (36 kodēšanas eksoni) “uz nākamās paaudzes sekvencēšanas paneļu analīzi”. JBTS4 gēns tiek pārbaudīts par homozigotu dzēšanu ar daudzkārtēju PCR. II posmā pārējie JB gēni tiek analizēti ar PCR (process, kas DNS ķēdē atkārto gēnu sekvences atkarībā no enzīma) un sekojošo Sangera sekvencēšanu atkarībā no fenotipa pazīmēm, kas atbilst mutāciju biežuma samazinājumam.
Lai izslēgtu hromosomu nelīdzsvarotību, tiek veikta diferenciāldiagnostikas SNP masīva analīze. Ja ir saskanīgums vai ja ģimenē ir zināmi vairāki slimi cilvēki, ārsti veic homozigotiskuma skrīningu, izmantojot savienojuma analīzi mikrosatellīta marķierā, kas apzīmē gēnu, un sekojošu gēna analīzi, izmantojot Sangera sekvencēšanu. No diagnostikas materiāla bērniem ņem divus līdz desmit mililitrus EDTA asiņu, no pieaugušajiem - no pieciem līdz desmit mililitriem.
Piemērots ir arī DNS vai audu materiāls. I posms: Ar genoma DNS materiālu pārbauda dublēšanos vai dzēšanu, izmantojot NPHP1 gēna kvantitatīvu analīzi, izmantojot MLPA. Ļoti nelielā daudzumā DNS genomā tiek pārbaudīti atsevišķu eksonu (gēnu segmentu) svītrojumi un dublēšanās. II posms: līdz šim identificēto gēnu kodētie eksoni tiek novērtēti, izmantojot nākamās paaudzes frekvences. Savienojumu vietas bagātina zondes hibridizācija.
Komplikācijas
Jouberta sindroms liek lielākajai daļai pacientu ciest no dažādām kaites. Tas parasti noved pie īsa auguma, elpošanas traucējumiem un, turklāt, ar palēnināšanos. Var ierobežot arī bērna garīgo attīstību. Elpošanas grūtības var izraisīt arī elpas trūkumu, kas noteikti jāārstē.
Nav neparasti, ka personas vecāki cieš no smagas depresijas vai citiem psiholoģiskiem traucējumiem. Pacientiem ir arī līdzsvara traucējumi un viņi bieži cieš no ierobežotas pārvietošanās spējas. Nav retums diskomforts acīm un ausīm, kas izraisa dzirdes zudumu vai redzes problēmas. Jouberta sindroms ievērojami samazina pacienta dzīves kvalitāti.
Ar dažādu terapiju palīdzību Jouberta sindromu var ierobežot un ārstēt. Diemžēl cēloņsakarību nevar veikt. Ārkārtas situācijās ārkārtas ventilāciju var veikt arī tad, ja trūkst elpas. Pašā ārstēšanā nav īpašu komplikāciju. Kopumā nevar paredzēt, vai pacienta dzīves ilgumu samazinās Jouberta sindroms.
Kad jāiet pie ārsta?
Grūtniecības laikā topošajai mātei jāpiedalās visās pieejamās pārbaudēs. Pārbaudēs tiek pārbaudīts grūtnieces, kā arī nedzimušā bērna veselības stāvoklis. Tā kā Džouberta sindromu var diagnosticēt jau 18. grūtniecības nedēļā, ieteicams izmantot profilaktiskās medicīniskās pārbaudes, ko iesaka veselības apdrošināšanas kompānijas. Turklāt, ja vecāku senču vēsturē ir kāds ģenētisks defekts, parasti ieteicams veikt ģenētiskas konsultācijas un izmeklēšanu.
Maz ticamā gadījumā, ja dzemdē netika konstatēti pārkāpumi, dzemdību speciālistu un pediatru automātiskas pārbaudes tiek veiktas tūlīt pēc dzemdībām. Šo izmeklējumu laikā var noteikt elpošanas traucējumus. Ja bērna vecāki pamana kādas neparastas neatbilstības, kuras iepriekš nav pamanītas, novērojumi jāapspriež ar ārstu. Ja ir kādas fiziskas īpatnības, īss augums vai deformācijas, jākonsultējas ar ārstu.
Ja, salīdzinot ar tāda paša vecuma bērniem, tiek pamanītas valodas problēmas vai garīga nepietiekama attīstība, jākonsultējas ar ārstu. Izmeklēšana ir nepieciešama, lai noskaidrotu cēloni. Jo ātrāk tiek noteikta diagnoze, jo agrāk var uzsākt mērķtiecīgu terapiju bērna uzturēšanai. Tāpēc pie pirmajām novirzes pazīmēm jākonsultējas ar ārstu.
Ārstēšana un terapija
Vecākiem ir tiesības uz ģenētiskām konsultācijām. Ārstēšanas iespējas ir dažādas, jo šīs slimības cēloņi ir dažādi. Motoriskas attīstības traucējumu un hipotensijas gadījumā tiek izmantotas izglītības atbalsta programmas, valodu, darba un ergoterapija, kas var labvēlīgi ietekmēt slimības gaitu.
Tiem, kurus skar neparasti elpošanas ieradumi, var veikt arī skābekļa aizvietošanu vai ventilāciju. Pacientiem ar viegliem simptomiem ir pozitīva prognoze. Ar smagi skartiem pacientiem jārūpējas ekspertu uzziņu centrā.
Perspektīva un prognoze
Jouberta sindroma prognoze ir slikta. Šis sindroms ir ģenētiska slimība. Ar pašreizējām medicīniskajām, zinātniskajām un juridiskajām prasībām to nevar izārstēt. Pētniekiem un ārstiem likumīgi nav atļauts mainīt cilvēka ģenētiskos apstākļus, izmantojot intervences. Šī iemesla dēļ ārstēšana ir vērsta uz tādu terapiju izmantošanu, kuru mērķis ir uzlabot esošo dzīves kvalitāti. Neizmantojot medicīnisko aprūpi, vēl vairāk samazinās pacienta labklājība.
Jo agrāk sindromu var diagnosticēt un ārstēt, jo labāki būs rezultāti. Ārkārtas situācijās ir norādīta attiecīgās personas ārkārtas ventilācija, pretējā gadījumā pacients var priekšlaicīgi nomirt. Lai arī daudzas terapijas ir apkopotas un piemērotas individuālā ārstēšanas plānā, esošā slimība var izraisīt sekundārus traucējumus. Tas pasliktina vispārējo prognozi.
Esošie funkcionālie traucējumi vai citi pārvietošanās ierobežojumi var izraisīt garīgas slimības. Daudziem pacientiem tiek dokumentēta īslaicīga vai pastāvīga depresija, garastāvokļa svārstības vai personības izmaiņas. Tas ir papildu slogs attiecīgajai personai un videi.Jouberta sindroma pacienta ikdienas dzīvi bieži var pārvaldīt tikai ar pietiekamu radinieku palīdzību un atbalstu. Līdzsvara traucējumi un ataksija ar vecumu kļūst smagāki.
novēršana
Tā kā precīza ģenētiskā cēloņsakarība vēl nav precīzi noteikta, klīniskajā nozīmē profilaktiski pasākumi nav veikti. Vienīgais veids, kā neitralizēt cilvēka organisma kroplības, ir veselīga dzīvesveida ievērošana.
Pēcaprūpe
Vairumā gadījumu pacientam ar Jouberta sindromu nav tiešu vai īpašu izsekošanas iespēju, tāpēc skartā persona galvenokārt ir atkarīga no ātras un, pats galvenais, agrīnas slimības diagnozes. Jo agrāk slimība tiek atzīta, jo labāks parasti būs tālākais kurss. Tāpēc pie pirmajiem simptomiem un pazīmēm ieteicams sazināties ar ārstu.
Ar šo slimību skartā persona parasti ir atkarīga no intensīvās aprūpes un terapijas, kas var mazināt simptomus. Ļoti nepieciešama arī vecāku un tuvu radinieku palīdzība un atbalsts, lai skartā persona varētu dzīvot normālu dzīvi. Bieži vingrinājumus no fizioterapijas vai fizioterapijas var veikt arī savās mājās, kas var mazināt simptomus.
Simptomus ne vienmēr var pilnībā mazināt. Ļoti noderīga var būt arī saziņa ar citiem Jouberta sindroma slimniekiem, jo informācijas apmaiņa nav nekas neparasts. Parasti šī slimība nesamazina skartās personas dzīves ilgumu.
To var izdarīt pats
Jouberta sindroms ir neārstējams, un arī ikdienas palīdzība ir grūta. Iedzimtas slimības simptomi vairumā gadījumu ir nenovēršami. Tomēr joprojām ir iespējams, ka daži no tiem tiks mazināti.
Tā kā elpošana ir īpaši traucēta skartajiem, tas ir sākumpunkts. Var būt noderīgs optimizēts istabas klimats. Sausais sildošais gaiss var saasināt elpošanas problēmas. Pārāk aukstam gaisam ir tāda pati ietekme. Ideālā gadījumā istabas temperatūra ir aptuveni 20 ° C un mitrums ap 50 procentiem. Īpaši istabas augi var veicināt optimālu iekštelpu klimatu. Alternatīvi telpā var ievietot arī mitrus dvieļus, lai mitrums būtu vēlamajā līmenī. Iekštelpu klimatu var izsekot, izmantojot higrometru. Vēl viens sākumpunkts, kas vērsts arī uz elpošanu, ir elpošanas vingrinājumi. Regulāra lietošana uzlabo uztveri par citādi automātisko procesu. Tādā veidā jūs varat novērst pārāk ātru elpošanu un elpošanas pauzes.
Tam ir jēga arī tad, ja skartie cilvēki istabā negulē vieni. Radinieki miega laikā var pamanīt elpošanas pārtraukumus un pamodināt pacientu vai stimulēt viņu elpot. Bet tā ir tikai piesardzība.
-durch-vitamin-b12-mangel.jpg)


.jpg)







.jpg)