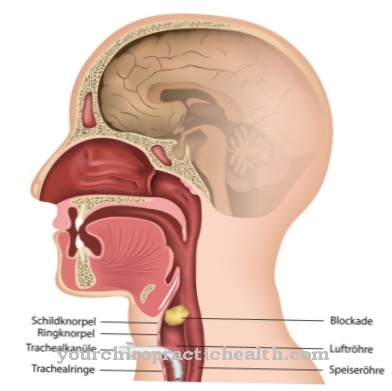
Kā De Toni Debre Fankoni sindroms sauc par ģenētisku slimību. Tas noved pie dažādu vielu reabsorbcijas nierēs.
Kas ir De Toni Debre Fanconi sindroms?

© kras99 - stock.adobe.com
Starp nosaukumiem ir arī De Toni Debre Fanconi sindroms De Toni Debre Fanconi komplekss, De Tonija Fankoni sindroms vai Glikozes aminoskābes diabēts zināms. Tiek domāts proksimālās kanāliņas nieru rezorbcijas traucējumi, kas veido nieru kanāliņu sekciju. De Toni Debre Fanconi sindroms ir iedzimta slimība, kuras rezultātā tiek absorbētas dažādas vielas.
Tajos ietilpst aminoskābes, glikoze un neorganiskais fosfors. Tas noved pie glikozes, fosfātu un aminoskābju patoloģiskas izdalīšanās. De Toni Debre Fanconi sindromu pirmo reizi aprakstīja itāļu ārsts Džovanni de Toni (1896-1973) un šveicietis Guido Fanconi (1892-1979), kuri arī šai slimībai deva savu nosaukumu.
De Toni Debre Fanconi sindroms ir viena no retāk sastopamajām slimībām. Eiropā ir zināmi apmēram 50 infantila tipa gadījumi. Tomēr sekundārās formas parādās biežāk.
cēloņi
De Toni Debre Fanconi sindromu var iedalīt trīs dažādās formās. Tas ir primārais idiopātiskais Fankoni sindroms, kas tiek mantots autosomāli recesīvā veidā, bet var rasties arī spontāni. Pieaugušo tipa Fanconi sindroms notiek mazāk smagi nekā infantilais tipa Fanconi sindroms.
Trešā forma ir sekundārais Fankoni sindroms. Tas notiek pavada iedzimtas vielmaiņas slimības, piemēram, cistinoze vai Lowe sindroms. Turklāt tas ir sekundārs Sjögrena sindroma, amiloidozes, audzēju, saindēšanās gadījumā vai kā zāļu nevēlama blakusparādība.
De Toni Debre Fanconi sindroms lielākoties ir iedzimta slimība. Kamēr sindroma infantilais tips tiek mantots kā autosomāli recesīva iezīme, pieaugušā tipa mantojums ir autosomāli dominējošs. De Toni Debre Fanconi sindroma gadījumā ir vispārēji proksimālās kanāliņas rezorbcijas traucējumi. Sindroma patomehānisms vēl nav pilnībā noskaidrots.
Tiek pieņemts, ka pastāv ar išēmiju saistīts ATP deficīts vai Na + - / K + ATPāzes nepietiekamība. Tā rezultātā dažādas vielas, piemēram, glikoze, aminoskābes vai joni, piemēram, fosfāts, vairs neizdalās un neizdalās no urīna. Tas, savukārt, izraisa aminoacidūriju, hiperkalciūriju, ieskaitot hipokaliēmiju, hiperfosfatūriju, kas saistīta ar fosfātu līdzsvara traucējumiem, un glikozūriju, ieskaitot osmotoisko diurēzi.
Papildus iedzimtajai De Toni Debre Fanconi sindroma formai, kas ir ģenētiska, ir arī iegūta forma. To izraisa vielmaiņas slimības, piemēram, fruktozes nepanesība vai Vilsona slimība, išēmija vai nefrotoksiskas vielas, piemēram, smagie metāli vai narkotikas.
Simptomi, kaites un pazīmes
De Toni Debre Fanconi sindroma simptomi ir atkarīgi no tā, vai tā ir infantila vai pieauguša cilvēka slimības forma. Idiopātiskā primārā sindroma gadījumā, kas rodas bērnībā, no 2 līdz 3 gadu vecumam parādās tādi simptomi kā īss augums, drudža lēkmes un vemšana. Turklāt slimie bērni cieš no rahīta, kas ir izturīgs pret D vitamīnu.
Ir arī stipras sāpes kaulos. Iespējami pat pārtraukumi. Ja nieru mazspēja, kas arī rodas, netiek ārstēta ar asiņu mazgāšanu (dialīzi) vai nieru transplantāciju, skartais bērns ir pat pakļauts nāves riskam.
Ja De Toni Debre Fanconi sindroma simptomi neparādās līdz pieauguša cilvēka vecumam, parasti nav jābaidās no dzīvībai bīstamām sekām. Pieaugušā sindroma forma kļūst pamanāma, pateicoties muskuļu hipotonijai, polidipsijai (patoloģiskām slāpēm) vai kaulu mīkstināšanai (osteomalācija).
Turklāt trūkuma dēļ pastāv komplikāciju risks. Tie ietver zemu cukura līmeni asinīs (hipoglikēmiju), spontānus lūzumus, neiroloģiskus traucējumus un hipokaliēmijas simptomus. Turklāt var rasties globāla nieru mazspēja.
Diagnostika un kurss
Lai diagnosticētu De Toni Debre Fanconi sindromu, ārsts, kurš veic pārbaudi, vispirms apskata pacienta slimības vēsturi. Viņš arī veic fizisko pārbaudi un medicīnisko laboratorisko pārbaudi. Balstoties uz urīna stāvokli, ir iespējams noteikt aminoacidūriju vai glikozūriju.
Multiplās mielomas gadījumā var noteikt proteinūriju. Fosfātu līmenis asins serumā ir samazināts. Reizēm var noteikt arī hipokaliēmiju. Lai diagnosticētu De Toni Debre Fanconi sindroma sekundāro efektu no osteomalācijas vai rahīta, var veikt rentgena starus. Dažreiz tiek veikta arī nieru biopsija (audu noņemšana).
De Toni Debre Fanconi sindroma gaita ir atkarīga no tā formas. Zīdaiņu formas prognoze tiek uzskatīta par nelabvēlīgu, jo bez nieru transplantācijas nieru mazspējas dēļ nāve iestājas no desmit līdz divpadsmit gadiem. Ja, no otras puses, tā ir pieaugušo forma, ko novēro tikai pieaugušajiem, dzīves ilgums ir normāls.
Kad jāiet pie ārsta?
Ja tiek novēroti tādi simptomi kā īss augums, drudža lēkmes un vemšana vecumā no diviem līdz trim gadiem, tas var būt De Toni-Debre-Fanconi sindroms. Apmeklējums pie pediatra ir ieteicams, ja simptomi neizzūd vienatnē vai ir papildu simptomi. Parasti jums vienmēr jāredz ārsts, ja jums ir kādas nopietnas slimības pazīmes. Smagu kaulu sāpju vai pat lūzumu gadījumā ir jābrīdina neatliekamās palīdzības dienesti vai cietušais bērns jānogādā slimnīcā.
Tas pats attiecas uz nieru mazspējas vai rahīta pazīmēm. Ja simptomi parādās tikai pieaugušā vecumā, ar patoloģiskām slāpēm, kaulu problēmām un citām raksturīgām pazīmēm jākonsultējas ar ārstu. Vēlākais komplikāciju, piemēram, hipoglikēmijas, neiroloģisku traucējumu vai spontānu lūzumu, gadījumā nepieciešama medicīniska konsultācija.
Papildus ģimenes ārstam var pieaicināt arī iedzimtu slimību speciālistu vai internistu. Nespecifisku simptomu gadījumā vislabāk ir vispirms sazināties ar neatliekamās medicīniskās palīdzības dienestu. Jebkurā gadījumā minētajām sūdzībām ir ieteicams medicīnisks precizējums.
Ārsti un terapeiti jūsu reģionā
Ārstēšana un terapija
Ārstējot De Toni Debre Fanconi sindromu, ir iespējama gan cēloņsakarība, gan simptomātiska terapija. Ja pacients cieš no sekundārās formas, tiek ārstēts primārās slimības cēlonis. Standarta terapija ietver D3 vitamīna vai kalcitriola piešķiršanu, lai ārstētu pret D vitamīnu izturīgus rahītus.
Hidrohlortiazīdu lieto arī, lai samazinātu elektrolītu zudumu caur nierēm. Noderīgas ir arī daudzas mazas maltītes, kurās ir daudz olbaltumvielu un ogļhidrātu. Turklāt pacientam dienā ir jāizdzer viens līdz trīs litri šķidruma. Tajā pašā laikā ir svarīgi samazināt galda sāls daudzumu. Ir svarīgi arī kompensēt fosfāta, kālija un nātrija zudumus.
Acidozi savukārt līdzsvaro buferšķīdumi. Ja ir iedzimti proksimālie traucējumi, ir iespējama tikai simptomātiska terapija, kurā pacients saņem nātrija bikarbonātu, fosfātu, glikozi un kāliju. Svarīga ir arī regulāra nieru un kaulu kontrole.
Perspektīva un prognoze
Tā kā De Toni Debre Fanconi sindroms ir ģenētiska slimība, to nevar ārstēt cēloņsakarībā, bet tikai simptomātiski. Pašdziedināšanās šajā sindromā nenotiek.
Ja to neārstē, De Toni Debre Fanconi sindroms var izraisīt pilnīgu nieru mazspēju, kas galu galā noved pie nāves. Pēc tam pacienti ir atkarīgi no donora nieres vai no dialīzes.
Sindroms var izraisīt arī īsu augumu, vemšanu vai dažādus deficīta simptomus. Dzīves kvalitāte un paredzamais dzīves ilgums ir ievērojami samazināts. Bērniem sindroms izraisa arī traucētu un palēninātu attīstību. Bieži ir kaulu lūzumi un brūču dzīšana.
De Toni Debre Fanconi sindromu parasti var relatīvi labi ārstēt ar medikamentu palīdzību. Tas atvieglo visus simptomus un noved pie pozitīvas slimības gaitas. Tomēr pacienti ir atkarīgi no šo zāļu lietošanas mūža garumā, jo sindromu nevar pilnībā izārstēt. Turklāt, lai izvairītos no komplikācijām, regulāri jāveic iekšējo orgānu pārbaudes. Ar agrīnu terapiju un veiksmīgu ārstēšanu dzīves ilgums nesamazinās.
novēršana
De Toni Debre Fanconi sindroms tiek pieskaitīts ģenētiskajām slimībām. Tāpēc nav preventīvu pasākumu.
Pēcaprūpe
De Toni Debre Fanconi sindroma gadījumā lielākajā daļā gadījumu skartajai personai ir tikai dažas iespējas tiešai sekojošai aprūpei. Lai izvairītos no turpmākām komplikācijām, skartajai personai savlaicīgi jāredz ārsts. Tā kā tā ir ģenētiska slimība, to nevar pilnībā izārstēt.
Ja vēlaties bērnus, ģenētiskās konsultācijas un testēšana var būt noderīga, lai novērstu slimības atkārtošanos. Pašdziedināšanās nenotiek De Toni Debre Fanconi sindroma gadījumā. Vairumā gadījumu pacientiem ir jālieto zāles pret šo slimību. Vienmēr ir svarīgi regulāri lietot pareizu devu.
Bērniem vecākiem ir jākontrolē uzņemšana. Arī skartajiem vajadzētu dzert daudz šķidruma un, ja iespējams, izvairīties no sāls, lai nevajadzīgi neradītu stresu nierēm. Tā kā De Toni Debre Fanconi sindroms var izraisīt smagus nieru darbības traucējumus, regulāra iekšējo orgānu kontrole un izmeklēšana ir ļoti svarīga. Īpaši jāpārbauda kauli un nieres. Nevar vispārīgi paredzēt, vai sindroma dēļ pacienta dzīves ilgums samazināsies.
To var izdarīt pats
Papildus ģenētiski noteiktajai iedzimtajai De Toni Debre Fanconi sindroma formai tas notiek arī iegūtā formā. Pats pacients nevar veikt nekādus pasākumus pret traucējumu ģenētisko variantu, kuriem ir cēloņsakarība.
Īpaši nopietna ir infantila slimības forma, kas parasti ir pamanāma starp otro un trešo dzīves gadu. Pēc tam vecākiem vajadzētu uzstāt, lai viņu bērnu ārstētu ārsts, kuram faktiski jau ir pieredze ar šo diezgan reti sastopamo slimību. Speciālisti var atrast caur ārstu asociāciju vai ar veselības apdrošināšanas kompānijas palīdzību.
Vienlaicīgi bieži sastopamā īsā auguma simptomus, piemēram, spriedzi, muskuļu sāpes vai ierobežotas motoriskās iemaņas, parasti var mazināt, ja fizioterapija tiek sākta agri. Tiklīdz bērni sāk emocionāli ciest no īsā auguma, jāaicina arī bērnu psihologs.
Ja slimība ir iegūta, nevis ģenētiska, pirmajam solim vajadzētu būt cēloņa identificēšanai. Tas var būt, piemēram, vielmaiņas slimība, fruktozes nepanesamība vai saindēšanās ar smagajiem metāliem. Zāles var būt atbildīgas arī par iegūto De Toni Debre Fanconi sindromu.
Ja slimība korelē ar pacienta dzīvesveidu, piemēram, uzturu vai nodarbošanos, viņam / viņai ir jāpielāgojas dzīvesveida izmaiņām.Ja nepieciešams mainīt uzturu, jākonsultējas ar dietologu. Ja šo profesiju vairs nevar praktizēt, agrīnā posmā jāmeklē nodarbinātības aģentūras karjeras konsultāciju dienests. Arī arodbiedrības šādos gadījumos konsultē bez maksas.


.jpg)
.jpg)






.jpg)