No Andersena slimība ir īpaši smaga glikogēna uzglabāšanas slimības forma.Tas ir iedzimts stāvoklis, kam raksturīga patoloģiska glikogēna veidošanās. Slimības prognoze ir ļoti slikta.
Kāda ir Andersena slimība?
© ag visuell - stock.adobe.com
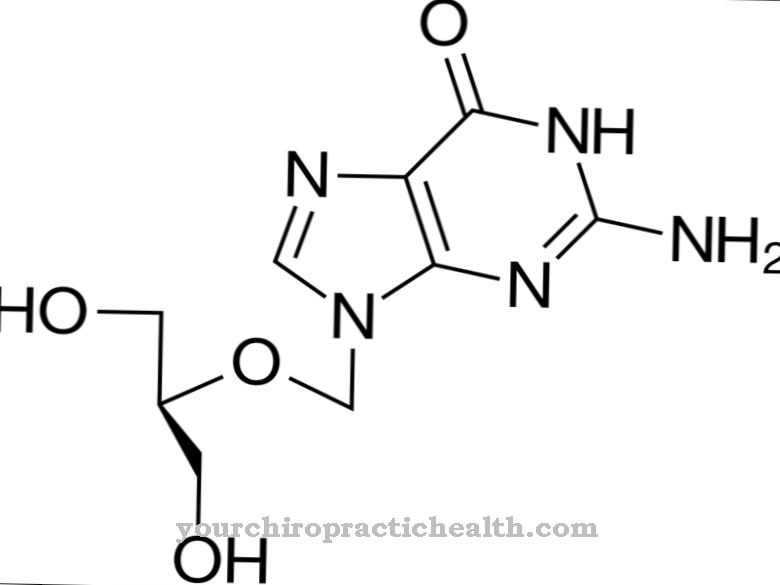
Kontekstā Andersena slimība tiek glabāta neparasta glikogēna forma. Šim glikogēnam ir līdzīga struktūra kā amilopektīnam, kura procentuālais daudzums ir atrodams dārzeņu ciete. Parasti glikogēns ir ļoti sazarots. Tomēr Andersena slimībā ir tikai vāji sazarots polisaharīds.
Slimību raksturo strauja aknu palielināšanās, kas ātri noved pie aknu cirozes. Nenormālo polisaharīdu vairs nevar sadalīt un turpina uzkrāties. Fermenta amilo-1,4-1,6-transglikozidāzes deficīts vai pat tā trūkums ir atbildīgs par nepilnīgu glikogēna veidošanos. Tas nodrošina šīs polisaharīdu molekulas sazarošanos.
Slimība ir ļoti reti sastopama, taču tā joprojām notiek dažādās formās vai formās. Īpaši smagā formā bērns bieži ir nedzīvs. Aprakstītas arī vieglākas formas, kas sākas vēlākā vecumā. Tomēr jebkurā gadījumā gēnā (GBE1), kas atrodas 3. hromosomā, ir mutācija.
cēloņi
Andersena slimības cēlonis ir ģenētisks defekts gēnā GBE1 3. hromosomā, ko var mantot kā autosomāli recesīvu iezīmi. Šis gēns ir atbildīgs par amil-1,4-1,6-transglikozidāzes fermenta sintēzi. Ja šī fermenta trūkst vai ja tam ir tikai ierobežota funkcionalitāte, parasto glikogēnu vairs nevar sintezēt. Ferments ir atbildīgs par polisaharozes molekulas sazarošanos.
Ja šī sazarošanās nenotiek vai ja tā tiek veikta tikai nepilnīgi, tiek izveidots glikogēns, kuru vairs nevar sadalīt ātrai enerģijas padevei. Gluži pretēji, tas ļoti ātri uzkrājas aknās, liesā un limfmezglos. Pēc katras ēdienreizes daļa neizlietotās glikozes tiek nogādāta aknās, lai to uzglabātu kā rezerves vielu - glikogēnu.
Tomēr šo rezerves materiālu nevar izmantot pašreizējā formā. Pastāvīga patoloģiskā glikogēna uzkrāšanās palielina aknas un liesu arvien vairāk un neizbēgami noved pie abu orgānu iznīcināšanas.
Simptomi, kaites un pazīmes
Andersena slimība izpaužas ar ārkārtēju mainīgumu. Runa ir par pastāvīgu nenormāla glikogēna glabāšanu, kuru vairs nav iespējams sadalīt. Bet slimības smagums var būt atšķirīgs. Neskatoties uz to, Andersena slimības prognoze kopumā ir ļoti slikta. Visredzamākais simptoms ir pastāvīgi palielinās aknas, no kurām ātri attīstās aknu ciroze.
Smagākā forma parāda sevi ar to, ka trūkst vai ir samazinātas bērna kustības pirms dzimšanas. Auglim ir locītavu stīvuma un plaušu hipoplāzijas pazīmes. Parasti šajos gadījumos bērns piedzimst miris. Klasiskajos gadījumos bērns parasti tiek attīstīts piedzimstot. Tomēr dažos pirmajos dzīves mēnešos attīstās hepatomegālija (palielinātas aknas) un hipotonija (muskuļu sasprindzinājuma trūkums).
Kopumā bērna attīstība tiek kavēta. Slimība strauji progresē. Aknās attīstās ciroze. Palielināts ir arī spiediens uz portālu un liesa palielinās. Sakarā ar aknu cirozi barības vadā attīstās variācijas ar atbilstošu asiņošanu un ascītu. Nāve parasti notiek agrā bērnībā. Retākos gadījumos slimība sākas vēlāk un parāda muskuļu vājuma un sirds mazspējas simptomus. Šeit rodas arī neiroloģiski simptomi.
Diagnoze un slimības gaita
Diagnozi var noteikt, pamatojoties uz klīnisko ainu, un tai var pievienot laboratorijas testus, aknu biopsijas un molekulārie ģenētiskos testus. Histoloģiskajos izmeklējumos ir pamanāma nemainīgu amilopektīnam līdzīgu struktūru uzkrāšanās intracelulāri. Atbildīgais enzīms tiek pārbaudīts hepatocītos, fibroblastos un leikocītos. Pierādīts amilo-1,4-1,6-transglikozidāzes deficīts apstiprina diagnozi.
Komplikācijas
Parasti Andersena slimība ievērojami samazina bērna dzīves ilgumu vai bērns piedzimst miris. Tas var izraisīt smagas psiholoģiskas sūdzības vai depresiju, īpaši ar radiniekiem vai vecākiem. Vairumā gadījumu viņi pēc tam ir atkarīgi no psiholoģiskas ārstēšanas.
Ietekmētie bērni cieš no aknu cirozes, kas galu galā noved pie nāves. Turklāt locītavas ir arī saīsinātas, un šīs sūdzības dēļ kustības vairs nav iespējamas. Bērna garīgo attīstību smagi pasliktina arī Andersena slimība, tāpēc skartie parasti vienmēr ir atkarīgi no citu cilvēku palīdzības. Nav nekas neparasts, ja rodas sirds mazspēja vai muskuļu vājums.
Pacienti var arī nomirt no sirds nāves. Diemžēl Andersena slimību nevar izārstēt. Aknu transplantācija simptomus var mazināt arī tikai uz īsu laiku, jo tiks nodarīts kaitējums arī jaunajām aknām. Tas galu galā noved pie bērna nāves. Tomēr līdz tam sūdzības un simptomus var ierobežot, izmantojot medicīniskus pasākumus.
Kad jāiet pie ārsta?
Andersena slimība ir ģenētiska slimība, kas smagos gadījumos var izraisīt augļa nāvi dzemdē. Tādēļ grūtniecēm jāmeklē medicīniskā palīdzība, tiklīdz grūtniecības laikā tiek pamanīti pārkāpumi vai novirzes no normas. Ja topošajai mātei ir neskaidra sajūta, ka ar nedzimušo bērnu kaut kas varētu būt nepareizi, viņai jākonsultējas ar ārstu. Ja jaundzimušais izdzīvo pirmās pāris dienas un nedēļas pēc piedzimšanas, ārsts ir nepieciešams, tiklīdz īpatnības kļūst redzamas turpmākajā attīstības gaitā. Ja Jums ir muskuļu vājums vai kustību traucējumi, jākonsultējas ar ārstu.
Augšanas traucējumi ir jau esošas slimības pazīmes, un tie ir jānoskaidro. Jāpārbauda un jāārstē sirds patoloģijas, ķermeņa deformācijas un atšķirības bērna uzvedībā. Daudzos gadījumos slimība noved pie orgānu palielināšanās. Šajos gadījumos tiek skartas jo īpaši aknas vai liesa.
Tādēļ ārsts ir nepieciešams, tiklīdz rodas neparasta ķermeņa augšdaļas forma, tieši salīdzinot ar zīdaiņiem vai tāda paša vecuma bērniem. Ādas krāsas maiņa vai citi ādas izskata pārkāpumi ir papildu veselības traucējumu pazīmes. Dzeltenīga seja vai acis jānovērtē ārstam.
Terapija un ārstēšana
Tā kā slimība ir ģenētiska, cēloņsakarību nevar veikt. Terapija ir tikai simptomātiska. Ārstēšanas laikā ārsti galvenokārt koncentrējas uz komplikācijām, kas rodas. Tas pazemina spiedienu portāla vēnu ķēdē. Ir arī albumīna un koagulācijas faktoru aizstāšana.
Ja rodas aknu mazspēja, aknu transplantācija var pagarināt dzīvi. Tomēr slimību nevar izārstēt pat ar aknu pārstādīšanu. Pastāv ģenētiskais defekts, un tas arī novedīs pie neparastā glikogēna uzkrāšanās jaunajās aknās. Bojātā polisaharīda glabāšana turpinās citos tā saucamās liesas un limfmezglu retikulohistiocitiskās sistēmas orgānos, lai arī pēc veiksmīgas aknu transplantācijas joprojām varētu rasties nopietnas komplikācijas.
Retikulohistiocītiskā sistēma ir imūnsistēmas sastāvdaļa un ietver tīklenes saistaudu šūnas. Šīs šūnas uzglabā daļiņas un vielas, lai tās sadalītu un pēc tam izvadītu no ķermeņa. Tomēr arī šeit vairs nav iespējama bojātu polisaharozes molekulu sadalīšana.
Perspektīva un prognoze
Andersena slimības prognoze ir salīdzinoši vāja. Metabolisma slimība līdz šim nav izārstējama un izraisa smagus aknu bojājumus. Dažos gadījumos rodas muskuļu sūdzības un vienlaicīgas slimības, kuras, ja tās neārstē, pakāpeniski progresē. Dzīves ilgumu nopietni ierobežo šis stāvoklis. Slimi bērni sasniedz vidēji divu līdz piecu gadu vecumu. Agrīna aknu transplantācija uzlabo prognozi. Prognoze ir slikta, īpaši attiecībā uz klasiskajām slimības formām, īpaši, ja pirmajos dzīves mēnešos nav aknu transplantācijas.
Parasti ilgtermiņa prognozes pamatā ir slimības pakāpe, smagums un progresēšana. Andersena slimība ir viena no smagākajām glikogenozēm. Dzīves kvalitāte parasti ievērojami pazeminās aknu darbības traucējumu un citu simptomu dēļ. Sāpju zāles un visaptveroša terapija uzlabo bērna labsajūtu, taču tie ir saistīti arī ar riskiem. Prognozi nodrošina atbildīgais aknu speciālists.
Dzīves ilgumu nopietni ierobežo šis stāvoklis. Prognozē iekļauj arī jebkādas vienlaicīgas slimības, kas var rasties ar neatklātām slimībām. Tādēļ Andersena slimības prognoze kopumā ir slikta. Jaunās ārstēšanas metodes nākotnē varētu dot uzlabojumus.
novēršana
Andersena slimības profilakse var attiekties tikai uz faktu, ka pēcnācēji šo slimību nemanto. Tā kā Andersena slimība tiek pārnesta autosomāli recesīvi, mantojumā var izlaist vairākas paaudzes. Ja ģimenē un radiniekos jau ir bijuši Andersena slimības gadījumi, tāpēc jāveic cilvēka ģenētiskā pārbaude.
Ja gēns ir atrasts abiem vecākiem, ieteicams konsultēt ģenētiski. Šajā gadījumā pēcnācējiem ir 25 procentu iespēja saslimt ar Andersena slimību.
Pēcaprūpe
Tā kā Andersena slimību nevar izārstēt, visa ārstēšanas perioda laikā galvenā uzmanība tiek pievērsta simptomu ārstēšanai un iespējamo komplikāciju ierobežošanai. Pēc iejaukšanās, kas tiek veikta kā terapijas daļa, nepieciešama papildu aprūpe. Ja notiek aknu transplantācija, tai ir ļoti svarīga profesionāla sekojoša aprūpe.
Pēc procedūras tas nodrošina, ka ķermenis neatmet jaunās aknas. Īpašas zāles nomāc ķermeņa imūno reakciju. Tomēr rezultātā tiek vājināta organisma izturība pret patogēniem, kas jāņem vērā turpmākajā terapijā. Šajā laikā pacientam regulāri jāveic asins analīzes. Rūpēs, lai nerastos atgrūšanas reakcijas vai citas nopietnas komplikācijas, piemēram, nieru disfunkcija, kas var rasties kā blakusparādības.
Lai arī Andersena slimības galvenos simptomus var uzlabot tūlīt pēc aknu transplantācijas, bojātā glikogēna nogulsnēšanās turpinās, tāpēc pat pēc transplantācijas ir sagaidāmas komplikācijas un progresējoši simptomi. Atbildīgais aknu speciālists var sniegt sīkāku informāciju par prognozi un turpmāko ārstēšanas gaitu.
To var izdarīt pats
Pašpalīdzības pasākumi, ko var veikt Andersena slimība pacients, ir ierobežoti. Tā kā slimībai ir ģenētiski cēloņi un to nevar kontrolēt, neskatoties uz simptomātisku ārstēšanu, skartās personas iespējas ātri tiek izsmeltas. Vislabāk ir ieteikt nopietni izturēties pret visiem ārstējošā ārsta ieteikumiem par uzturu un dzīvesveidu un tos ieviest.
Turklāt pēc aknu transplantācijas skartajai personai jāapsver saudzīga izturēšanās. Jāizvairās no alkohola, taukainas pārtikas un piepūles. Tas ķermenim ļauj vieglāk pieņemt jauno orgānu. Tomēr veiksmīga transplantācija, ieskaitot veiksmīgu turpmāko aprūpi, pati par sevi nevar apturēt 4. tipa glikogenozi.
Tā kā šī ir autosomāli recesīva iedzimta slimība (tā var izlaist vairākas paaudzes), plānojot ģimeni, ir jēga sastādīt ģenētisko profilu. Kaut arī tie, kurus skārusi Andersena slimība, jau zina par savu gēnu, analīze šajā sakarā ir īpaši vērtīga ģimenes locekļiem. Šādā veidā ierosinošā gēna pārnešanu var novērst, veicot atbilstošu ģimenes plānošanu. Vismaz tomēr var iegūt skaidrību par slimības risku pēcnācējiem.

.jpg)
.jpg)


.jpg)